
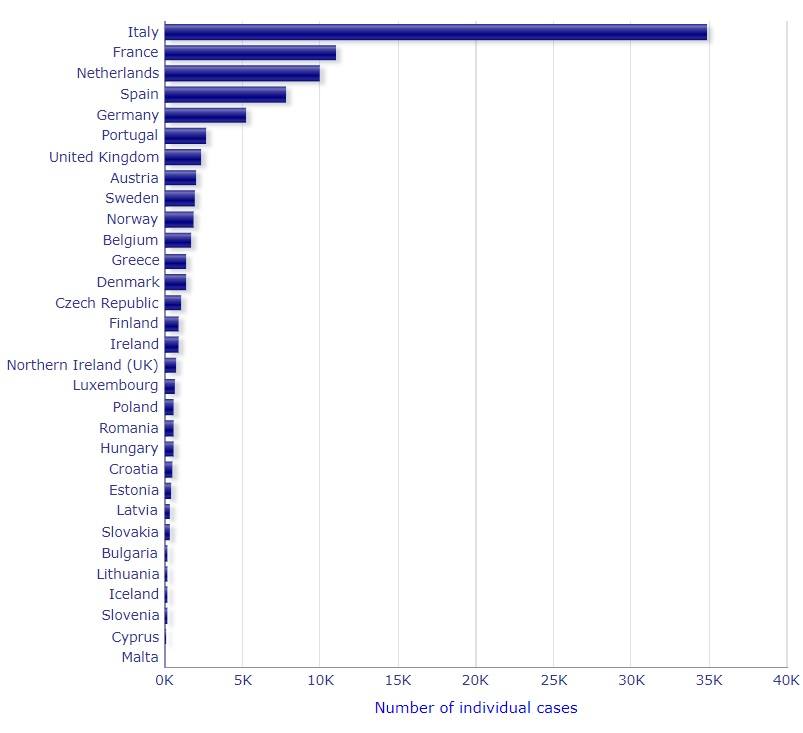
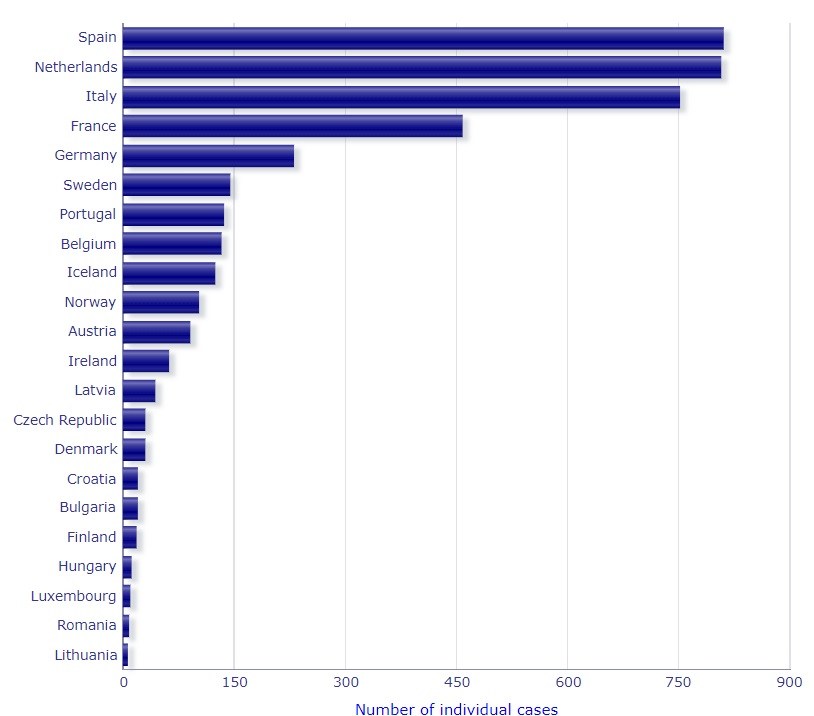
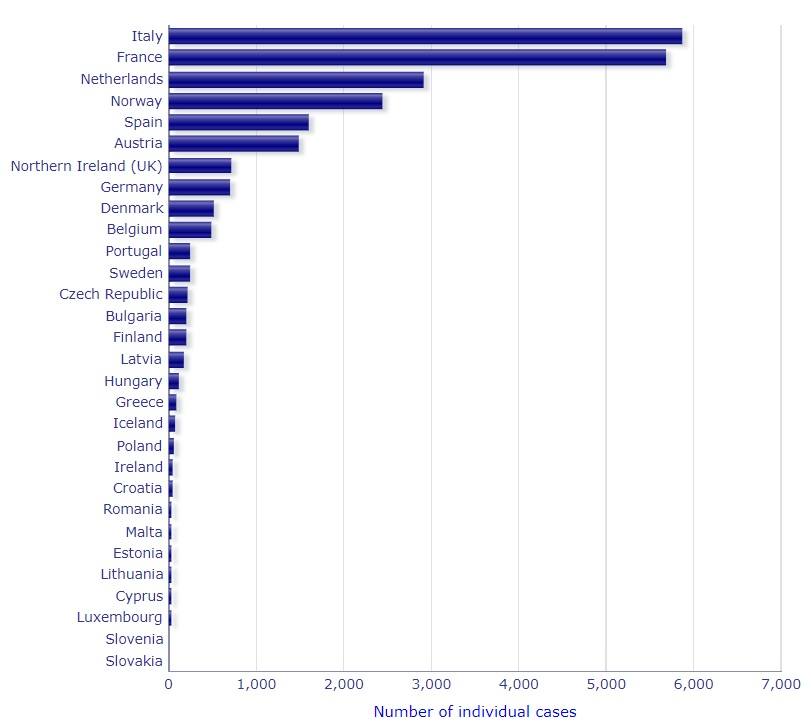
COVID-19: Meldung von Nebenwirkungen von Arzneimitteln an die EMA
MEDIKAMENTE FÜR KINDER
EU- Arbeitsteilung bei der Auswertung pädiatrischer Daten
EMA,-Das Fehlen geeigneter Informationen für viele zugelassene Arzneimittel zur Behandlung von Erkrankungen bei Kindern gibt seit einiger Zeit Anlass zur Sorge. Mehr als 50 % der Arzneimittel, die in Europa zur Behandlung von Kindern verwendet werden, wurden nicht für die Anwendung bei Kindern getestet und sind für Kinder nicht zugelassen.
In jüngster Zeit sind aufgrund der Initiative in den USA, die pharmazeutische Industrie zur Durchführung klinischer Studien an Kindern anzuregen, über Medikamente, die bereits für die erwachsene Bevölkerung verwendet werden, mehr Informationen verfügbar. Auch in der EUeine solche Initiative wurde von der Europäischen Kommission ergriffen, aber diese neue pädiatrische Verordnung wird erst Ende 2006 oder Anfang 2007 in Kraft treten und es wird einige Zeit dauern, bis infolge der pädiatrischen Verordnung mehr Arzneimittel für Kinder verfügbar sein werden. Darüber hinaus werden die meisten zukünftigen Studien an Kindern mit neuen Arzneimitteln durchgeführt.
Für bereits auf dem Markt befindliche Arzneimittel forderten die zuständigen nationalen Behörden die Zulassungsinhaber auf, alle Daten zur Anwendung von Arzneimitteln bei Kindern einzureichen. Die zuständigen nationalen Behörden arbeiten bei der Auswertung der pädiatrischen Daten mit der Absicht zusammen, sich auf die gleichen Informationen zu einigen und die Arbeitsbelastung zu teilen. Diese Initiative heißt EUArbeitsteilungsverfahren bei der Auswertung pädiatrischer Daten. Das Hauptprinzip besteht darin, dass zwei Mitgliedstaaten die Daten auswerten und einen Bewertungsbericht für die anderen Mitgliedstaaten erstellen. In einem vereinbarten Zeitrahmen können andere Mitgliedstaaten zu den Bewertungsberichten Stellung nehmen.
Die Schlussfolgerungen aus der Bewertung werden in die Medizinproduktinformationen aufgenommen und die Bewertungsberichte werden veröffentlicht, um den Angehörigen der Gesundheitsberufe mehr Informationen zur Verfügung zu stellen.
Obwohl die neu eingereichten Daten meistens nicht ausreichen, um eine Indikation für die Anwendung bei Kindern zu genehmigen, können die verfügbaren Daten nützliche und wichtige Informationen für Angehörige der Gesundheitsberufe liefern, die entscheiden müssen, welche Arzneimittel zur Behandlung von Kindern verwendet werden können.
Weitere Einzelheiten zum Arbeitsteilungsverfahren, wie es von den Leitern der Arzneimittelbehörden ( HMA ) vereinbart wurde , finden Sie im Best Practice Guide zum EU- Arbeitsteilungsverfahren bei der Bewertung von pädiatrischen Daten.
Dieses Arbeitsteilungsverfahren ist ein laufendes Projekt und neue Bewertungsberichte werden hinzugefügt, wenn die Verfahren abgeschlossen sind.
Februar 2021 CMDh/429/2021
Zusammenfassung des Risikomanagementplans für Name(n)> (Dexamethason) zur Behandlung des Coronavirus
Krankheit 2019 (COVID-19)
Dies ist eine Zusammenfassung des Risikomanagementplans (RMP) für .
Die RMP-Details wichtige Risiken von , wie diese Risiken minimiert werden können und wie weitere Informationen
über die Risiken und Unsicherheiten von eingeholt werden (fehlende Informationen).
Die Zusammenfassung der Merkmale des Arzneimittels (SmPC) und die Packungsbeilage von geben wesentliche
Informationen für medizinisches Fachpersonal und Patienten zur Anwendung von .
Wichtige neue Bedenken oder Änderungen an den aktuellen werden in die Updates von s RMP.
I. Das Arzneimittel und wofür es angewendet wird
ist für zugelassen (siehe SmPC für die vollständige Indikation). Es beinhaltet
Dexamethason als Wirkstoff und wird auf verabreicht.
II. Risiken im Zusammenhang mit dem Arzneimittel und Aktivitäten zur
Risiken minimieren oder weiter charakterisieren
Wichtige Risiken von , zusammen mit Maßnahmen zur Minimierung solcher Risiken und den vorgeschlagenen
Studien, um mehr über die Risiken von zu erfahren, werden im Folgenden beschrieben.
Maßnahmen zur Minimierung der identifizierten Risiken für Arzneimittel können sein:
• Spezifische Informationen, wie Warnhinweise, Vorsichtsmaßnahmen und Ratschläge zur korrekten Anwendung, in der Packung
Packungsbeilage und Fachinformation für Patienten und medizinisches Fachpersonal;
• Wichtige Hinweise auf der Verpackung des Arzneimittels;
• Die zugelassene Packungsgröße – die Arzneimittelmenge in einer Packung wird so gewählt, dass die
das Medikament wird richtig angewendet;
Zusammenfassung des Risikomanagementplans für (Dexamethason) für
Behandlung der Coronavirus-Krankheit 2019 (COVID-19)
Seite 2/3
• Rechtsstatus des Arzneimittels – die Art und Weise, wie ein Arzneimittel an den Patienten geliefert wird (z. B. mit oder ohne Rezept) kann dazu beitragen, die Risiken zu minimieren.
Zusammen bilden diese Maßnahmen routinemäßige Maßnahmen zur Risikominimierung.
Zusätzlich zu diesen Maßnahmen werden kontinuierlich Informationen über Nebenwirkungen gesammelt und
regelmäßig analysiert <, einschließlich PSUR-Beurteilung,> damit sofort Maßnahmen ergriffen werden können
notwendig. Diese Maßnahmen stellen routinemäßige Pharmakovigilanz-Aktivitäten dar.
Wenn wichtige Informationen, die die sichere Anwendung von beeinflussen könnten, noch nicht verfügbar sind, ist es
unten unter „fehlende Informationen“ aufgeführt.
II.A Liste wichtiger Risiken und fehlender Informationen
Wichtige Risiken von sind Risiken, die besondere Risikomanagementaktivitäten erfordern, um weiter
das Risiko untersuchen oder minimieren, damit das Arzneimittel sicher werden kann.
Wichtige Risiken können als identifiziert oder potenziell angesehen werden. Identifizierte Risiken sind Bedenken, für die es ist ein ausreichender Nachweis für eine Verknüpfung mit der Verwendung von . Potentielle Risiken sind Bedenken, für die ein Ein Zusammenhang mit der Anwendung dieses Arzneimittels ist auf Grundlage der verfügbaren Daten möglich, aber dieser Zusammenhang hat noch nicht festgelegt und bedarf weiterer Evaluierung. Fehlende Angaben beziehen sich auf Angaben zu den Sicherheit des Arzneimittels, das derzeit fehlt und erfasst werden muss (z. B. bei der Langzeitanwendung des Arzneimittels);
Liste wichtiger Risiken und fehlender Informationen
Wichtige identifizierte Risiken • Keine
Wichtige potenzielle Risiken • Keine
Fehlende Informationen • Sicherheit bei Patienten >70 Jahre und insbesondere >80
Jahre alt (COVID-19-Indikation)
• Sicherheit bei Schwangeren (COVID-19-Indikation)
II.B Zusammenfassung wichtiger Risiken und fehlender Informationen
Fehlende Informationen: Sicherheit bei Patienten >70 Jahre und insbesondere >80 Jahre (COVID-19
Indikation)
Maßnahmen zur Risikominimierung Routinemäßige Maßnahmen zur Risikominimierung:
SmPC-Abschnitt 5.1
Zusätzliche Maßnahmen zur Risikominimierung:
Keiner
Fehlende Informationen: Sicherheit bei Schwangeren (COVID-19-Indikation)
Maßnahmen zur Risikominimierung Routinemäßige Maßnahmen zur Risikominimierung:
SmPC Abschnitt 4.6 und PL Abschnitt 2
Zusätzliche Maßnahmen zur Risikominimierung:
Keiner
Zusammenfassung des Risikomanagementplans für (Dexamethason) für
Behandlung der Coronavirus-Krankheit 2019 (COVID-19)
Seite 3/3
II.C Entwicklungsplan nach der Zulassung
II.C.1 Studien, die Bedingungen der Genehmigung für das Inverkehrbringen sind
Es liegen keine Studien vor, die Bedingungen der Genehmigung für das Inverkehrbringen oder besondere Verpflichtung von
.
II.C.2 Sonstige Studien in Post-Autor
Wichtige hinweise Wenn sie Ein Nebenwirkung melden folgen sie unten Information hinweise
So melden Sie eine Nebenwirkung
Die Meldung von Nebenwirkungen erfolgt normalerweise durch medizinisches Fachpersonal , und es wird daher empfohlen, mit einem medizinischen Fachpersonal wie Ihrem Arzt oder Apotheker zu sprechen.
Patienten können vermutete Nebenwirkungen auch direkt über verschiedene Methoden melden, z. B. über Online-Meldeformulare für Patienten, die von den nationalen Arzneimittelzulassungsbehörden bereitgestellt werden, oder telefonisch. Informationen zur Meldung einer Nebenwirkung erhalten Sie bei der zuständigen Behörde aus der Liste der nationalen Arzneimittelzulassungsbehörden im EWR .
Wenn Sie eine Nebenwirkung bemerken oder vermuten, dass Sie eine solche haben, sollten Sie sich von einem Arzt beraten lassen . Die Europäische Arzneimittel-Agentur kann keine Berichte über Nebenwirkungen direkt von Patienten oder Verbrauchern akzeptieren. Die Agentur ist auch nicht in der Lage, eine individuelle medizinische Beratung zu erteilen oder zu bestätigen, ob Ihre Symptome durch Ihr Arzneimittel verursacht werden.
Weitere Informationen: Wussten Sie schon? Nebenwirkungen können Sie selbst melden http://www.adrreports.eu/en/report_side_effect.html/https://www.hma.eu/621.html



You must be logged in to post a comment.